
3051A - 1 Revision 1
February 2007
METHOD 3051A
MICROWAVE ASSISTED ACID DIGESTION OF
SEDIMENTS, SLUDGES, SOILS, AND OILS
SW-846 is not intended to be an analytical training manual. Therefore, method
procedures are written based on the assumption that they will be performed by analysts who are
formally trained in at least the basic principles of chemical analysis and in the use of the subject
technology.
In addition, SW-846 methods, with the exception of required method use for the analysis
of method-defined parameters, are intended to be guidance methods which contain general
information on how to perform an analytical procedure or technique which a laboratory can use
as a basic starting point for generating its own detailed Standard Operating Procedure (SOP),
either for its own general use or for a specific project application. The performance data
included in this method are for guidance purposes only, and are not intended to be and must
not be used as absolute QC acceptance criteria for purposes of laboratory accreditation.
1.0 SCOPE AND APPLICATION
1.1 This microwave extraction method is designed to mimic extraction using
conventional heating with nitric acid (HNO
3
), or alternatively, nitric acid and hydrochloric acid
(HCl), according to EPA Method 200.2 and Method 3050. Since this method is not intended to
accomplish total decomposition of the sample, the extracted analyte concentrations may not
reflect the total content in the sample. This method is applicable to the microwave-assisted acid
extraction/dissolution
‡
of sediments, sludges, soils, and oils for the following elements:
Element CAS Registry No.
a
*Aluminum (Al) 7429-90-5
*Antimony (Sb) 7440-36-0
Arsenic (As) 7440-38-2
*Barium (Ba) 7440-39-3
*Beryllium (Be) 7440-41-7
Boron (B) 7440-42-8
Cadmium (Cd) 7440-43-9
Calcium (Ca) 7440-70-2
*Chromium (Cr) 7440-47-3
Cobalt (Co) 7440-48-4
Copper (Cu) 7440-50-8
*Iron (Fe) 7439-89-6

Element CAS Registry No.
a
3051A - 2 Revision 1
February 2007
Lead (Pb) 7439-92-1
*Magnesium (Mg) 7439-95-4
Manganese (Mn) 7439-96-5
Mercury (Hg) 7439-97-6
Molybdenum (Mo) 7439-98-7
Nickel (Ni) 7440-02-0
Potassium (K) 7440-09-7
Selenium (Se) 7782-49-2
*Silver (Ag) 7440-22-4
Sodium (Na) 7440-23-5
Strontium (Sr) 7440-24-6
Thallium (Tl) 7440-28-0
*Vanadium (V) 7440-62-2
Zinc (Zn) 7440-66-6
a
Chemical Abstract Service Registry Number
*Indicates elements which typically require the addition of HCl to achieve
equivalent results with Method 3050, as noted in Ref. 3.
‡
Note: For matrices such as certain types of oils, this method may or may not
provide total sample dissolution. For other matrices, such as soils and sediments,
it should be considered an extraction method. Other elements and matrices may
be analyzed by this method if performance is demonstrated
for the analyte of
interest, in the matrices of interest, at the concentration levels of interest (see
Sec. 9.0).
1.2 This method is provided as an alternative to EPA Method 200.2 and Method 3050.
This method provides options for improving the performance for certain analytes, such as
antimony, iron, aluminum, and silver by the addition of hydrochloric acid, when necessary. It is
intended to provide a rapid multi-element acid extraction or dissolution prior to analysis so that
decisions can be made about materials and site cleanup levels, the need for TCLP testing of a
waste (see Method 1311), and whether a BDAT process is providing acceptable performance.
Digests produced by the method are suitable for analysis by flame atomic absorption
spectrophotometry (FLAA), graphite furnace atomic absorption spectrophotometry (GFAA),
inductively coupled plasma atomic emission spectrometry (ICP-AES) and inductively coupled
plasma mass spectrometry (ICP-MS). However, the addition of HCl may limit the quantitation
methods, or increase the difficulties of quantitation with some techniques.
Due to the rapid advances in microwave technology, consult your manufacturer's
recommended instructions for guidance on their microwave digestion system.
3051A - 3 Revision 1
February 2007
1.3 Prior to employing this method, analysts are advised to consult the determinative
method that may be employed in the overall analysis for additional information on quality control
procedures, development of QC acceptance criteria, calculations, and general guidance.
Analysts also should consult the disclaimer statement at the front of the manual and the
information in Chapter Two for guidance on the intended flexibility in the choice of methods,
apparatus, materials, reagents, and supplies, and on the responsibilities of the analyst for
demonstrating that the techniques employed are appropriate for the analytes of interest, in the
matrix of interest, and at the levels of concern.
In addition, analysts and data users are advised that, except where explicitly specified in a
regulation, the use of SW-846 methods is not mandatory in response to Federal testing
requirements. The information contained in this method is provided by EPA as guidance to be
used by the analyst and the regulated community in making judgments necessary to generate
results that meet the data quality objectives for the intended application.
1.4 Use of this method is restricted to use by, or under supervision of, properly
personnel experienced and trained in the use of microwave digestion systems. Each analyst
must demonstrate the ability to generate acceptable results with this method.
2.0 SUMMARY OF METHOD
A representative sample is extracted and/or dissolved in concentrated nitric acid, or
alternatively, concentrated nitric acid and concentrated hydrochloric acid using microwave
heating with a suitable laboratory microwave unit. The sample and acid(s) are placed in a
fluorocarbon polymer (PFA or TFM) or quartz microwave vessel or vessel liner. The vessel is
sealed and heated in the microwave unit for a specified period of time. After cooling, the vessel
contents are filtered, centrifuged, or allowed to settle and then diluted to volume and analyzed
by the appropriate determinative method.
3.0 DEFINITIONS
Refer to Chapter One, Chapter Three and the manufacturer's instructions for definitions
that may be relevant to this procedure.
4.0 INTERFERENCES
4.1 Solvents, reagents, glassware, and other sample processing hardware may yield
artifacts and/or interferences to sample analysis. All of these materials must be demonstrated
to be free from interferences under the conditions of the analysis by analyzing method blanks.
Specific selection of reagents and purification of solvents by distillation in all-glass systems may
be necessary. Refer to each method to be used for specific guidance on quality control
procedures and to Chapter Three for general guidance on the cleaning of glassware. Also refer
to the determinative methods to be used for a discussion of interferences.
4.2 Very reactive samples or volatile materials may create high pressures due to the
evolution of gaseous digestion products. This may cause venting of the vessels with potential
loss of sample and/or analytes. The complete decomposition of either carbonates, or carbon
based samples, may produce enough pressure to vent the vessel if the sample size is greater
than 0.25 g (depending on the pressure capability of the vessel). Variations of the method to
accommodate very reactive materials are specifically addressed in Sec. 11.3.3.

3051A - 4 Revision 1
February 2007
4.3 Many types of samples will be dissolved by this method. A few refractory sample
matrix compounds, such as quartz, silicates, titanium dioxide, alumina, and other oxides may
not be dissolved and in some cases may sequester target analyte elements. These bound
elements are considered non-mobile in the environment and are excluded from most aqueous
transport mechanisms of pollution.
4.4 Samples that are highly reactive or contaminated may require dilution before
analysis. If samples are diluted, then any dilutions must be accounted for in all subsequent
calculations. Highly reactive samples may also require pre-digestion in a hood to minimize the
danger of thermal runaway and excessively vigorous reactions.
5.0 SAFETY
5.1 This method does not address all safety issues associated with its use. The
laboratory is responsible for maintaining a safe work environment and a current awareness file
of OSHA regulations regarding the safe handling of the chemicals listed in this method. A
reference file of material safety data sheets (MSDSs) should be available to all personnel
involved in these analyses.
5.2 The microwave unit cavity must be corrosion resistant and well ventilated. All
electronics must be protected against corrosion for safe operation.
CAUTION
: There are many safety and operational recommendations specific to the model and
manufacturer of the microwave equipment used in individual laboratories. A listing
of these specific suggestions is beyond the scope of this method. The analyst is
advised to consult the equipment manual, the equipment manufacturer, and other
appropriate literature for proper and safe operation of the microwave equipment
and vessels. For further details and a review of safety methods during microwave
sample preparation, see Ref. 3 and the document of Sec. 13.3.1.
5.3 This method requires microwave transparent and reagent resistant materials such
as fluorocarbon polymers (examples are PFA or TFM) or quartz to contain acids and samples.
For higher pressure capabilities the vessel may be contained within layers of different
microwave transparent materials for strength, durability, and safety. The internal volume of the
vessel should be at least 45 mL, and the vessel must be capable of withstanding pressures of at
least 30 atm (435 psi), and capable of controlled pressure relief. These specifications are to
provide an appropriate, safe, and durable reaction vessel of which there are many adequate
designs by many suppliers.
WARNING
: The reagent combination (9 mL nitric acid to 3 mL hydrochloric acid) results in
greater pressures than those resulting from the use of only nitric acid. As
demonstrated in Figures 1 and 2, pressures of approximately 12 atm have been
reached during the heating of the acid mixture alone (no sample in the vessel).
Pressures reached during the actual decomposition of a sediment sample (SRM
2704, a matrix with low organic content) have more than doubled when using the 9
mL nitric and 3 mL hydrochloric acid mixture. These higher pressures necessitate
the use of vessels having higher pressure capabilities (30 atm or 435 psi).
Matrices having large organic content, such as oils, can produce approximately 25
atm of pressure inside the vessel (as described in Method 3052).

3051A - 5 Revision 1
February 2007
WARNING
: The outer layers of vessels are frequently not as acid or reagent resistant as the
liner material. In order to retain the specified performance and safety
requirements, these outer layers must not be chemically degraded or physically
damaged. Routine examination of the vessel materials is necessary to ensure
their safe use.
WARNING
: Another safety concern relates to the use of sealed containers without pressure
relief devices. Temperature is the important variable controlling the reaction.
Pressure is needed to attain elevated temperatures, but must be safely contained.
Some digestion vessels constructed from certain fluorocarbons may crack, burst,
or explode in the unit under certain pressures. Only vessels approved by the
manufacturer of the microwave system being used are considered acceptable.
WARNING
: When acids such as nitric and hydrochloric are used to effect sample digestion in
microwave units in open vessel(s), or sealed vessel(s), there is the potential for
any released acid vapors to corrode the safety devices that prevent the microwave
magnetron from shutting off when the door is opened. This can result in operator
exposure to microwave energy. Use of a laboratory-grade microwave equipment
system with isolated and corrosion resistant safety devices prevents this from
occurring. Use of laboratory-grade microwave equipment is needed to prevent
safety hazards. For further details, consult Ref. 3 and the document listed in Sec.
13.3.1.
Users are therefore advised not to use domestic (kitchen) type microwave ovens or
sealed containers which are not equipped with controlled pressure relief
mechanisms for microwave acid digestions by this method.
6.0 EQUIPMENT AND SUPPLIES
The mention of trade names or commercial products in this manual is for illustrative
purposes only, and does not constitute an EPA endorsement or exclusive recommendation for
use. The products and instrument settings cited in SW-846 methods represent those products
and settings used during method development or subsequently evaluated by the Agency.
Glassware, reagents, supplies, equipment, and settings other than those listed in this manual
may be employed provided that method performance appropriate for the intended application
has been demonstrated and documented.
This section does not list common laboratory glassware (e.g., beakers and flasks).
6.1 Microwave apparatus requirements
6.1.1 The temperature performance requirements necessitate the microwave
decomposition system to sense the temperature to within ± 2.5 EC and automatically
adjust the microwave field output power within 2 seconds of sensing. Temperature
sensors should be accurate to ± 2 EC (including the final reaction temperature of 175 ± 5
EC). Temperature feedback control provides the primary performance mechanism for the
method. Due to the variability in sample matrix types and microwave digestion equipment
(i.e., different vessel types and microwave oven designs), temperature feedback control is
preferred for reproducible microwave heating. For further details consult Ref. 3.
Alternatively, for a specific vessel type, specific set of reagent(s), and sample type,
a calibration control mechanism can be developed. Through calibration of the microwave
3051A - 6 Revision 1
February 2007
power for a specific number and type of vessels, vessel load, and heat loss characteristics
of a specific vessel series, the reaction temperature profile described in Sec. 11.3.5 can
be reproduced. The calibration settings are specific for the number and type of vessels
and microwave system being used, in addition to the specific reagent combination being
used. Therefore, no specific calibration settings are provided in this method. These
settings may be developed by using temperature monitoring equipment for each specific
set of microwave equipment and vessel type. They may be used as previously described
in such methods as Methods 3015 and 3052. In this circumstance, the microwave system
provides programmable power, which can be programmed to within ± 12 W of the required
power. Typical systems provide a nominal 600 W to 1200 W of power. Calibration control
provides backward compatibility with older laboratory microwave systems which may not
be equipped for temperature monitoring or feedback control and with lower cost
microwave systems for some repetitive analyses. Older vessels with lower pressure
capabilities may not be compatible (see Refs. 1, 2, and 3 and the documents listed in
Secs. 13.3.3 and 13.3.5).
6.1.2 The accuracy of the temperature measurement system should be
periodically validated at an elevated temperature. This can be done using a container of
silicon oil (a high temperature oil) and an external, calibrated temperature measurement
system. The oil should be adequately stirred to ensure a homogeneous temperature, and
both the microwave temperature sensor and the external temperature sensor placed into
the oil. After heating the oil to a constant temperature of 180 ± 5 EC, the temperature
should be measured using both sensors. If the measured temperatures vary by more than
1 to 2 EC, the microwave temperature measurement system should be calibrated. Consult
the microwave manufacturer’s instructions about the specific temperature sensor
calibration procedure.
6.1.3 A rotating turntable is employed to ensure the homogeneous distribution
of microwave radiation within the unit. The speed of the turntable should be a minimum of
3 rpm. Other types of equipment that are used to assist in achieving uniformity of the
microwave field may also be appropriate.
6.2 Filter paper, qualitative or equivalent.
6.3 Filter funnel, glass, polypropylene, or other appropriate material.
6.4 Analytical balance, of appropriate capacity and resolution meeting data quality
objectives.
7.0 REAGENTS AND STANDARDS
7.1 Reagent-grade chemicals must be used in all tests. Unless otherwise indicated, it
is intended that all reagents conform to the specifications of the Committee on Analytical
Reagents of the American Chemical Society, where such specifications are available. Other
grades may be used, provided it is first ascertained that the reagent is of sufficiently high purity
to permit its use without lessening the accuracy of the determination.
7.2 All acids should be sub-boiling distilled where possible to minimize the blank levels
due to metallic contamination. Other grades may be used, provided it is first ascertained that
the reagent is of sufficient purity to permit its use without decreasing the accuracy of the
determination. If the purity of a reagent is questionable, the reagent should be analyzed to
3051A - 7 Revision 1
February 2007
determine the level of impurities. The reagent blank must be less than the lower level of
quantitation in order to be used.
7.2.1 Concentrated nitric acid (HNO
3
) -- The acid should be analyzed to
determine levels of impurity. If the method blank is less than the lower level of
quantitation, the acid can be used.
7.2.2 Concentrated hydrochloric acid (HCl) -- The acid should be analyzed to
determine levels of impurity. If the method blank is less than the lower level of
quantitation, the acid can be used.
7.3 Reagent water -- Reagent water must be interference free. All references to water
in this method refer to reagent water unless otherwise specified. For further details, consult the
document listed in Sec. 13.3.2.
8.0 SAMPLE COLLECTION, PRESERVATION, AND STORAGE
8.1 See the introductory material to Chapter Three, "Inorganic Analytes."
8.2 All sample containers must be prewashed with acids and water, and metal-free
detergents, if necessary, depending on the history of use of the container (Ref. 3). Plastic and
glass containers are both suitable. For further information, see Chapter Three.
9.0 QUALITY CONTROL
9.1 Refer to Chapter One for additional guidance on quality assurance (QA) and
quality control (QC) protocols. When inconsistencies exist between QC guidelines, method-
specific QC criteria take precedence over both technique-specific criteria and those criteria
given in Chapter One, and technique-specific QC criteria take precedence over the criteria in
Chapter One. Any effort involving the collection of analytical data should include development
of a structured and systematic planning document, such as a Quality Assurance Project Plan
(QAPP) or a Sampling and Analysis Plan (SAP), which translates project objectives and
specifications into directions for those that will implement the project and assess the results.
Each laboratory should maintain a formal quality assurance program. The laboratory should
also maintain records to document the quality of the data generated. All data sheets and quality
control data should be maintained for reference or inspection.
9.2 Initial demonstration of proficiency
Each laboratory must demonstrate initial proficiency with each sample preparation and
determinative method combination it utilizes by generating data of acceptable accuracy and
precision for target analytes in a clean reference matrix. This will include a combination of the
sample extraction method and the determinative method (a 6000 or 7000 series method). The
laboratory must also repeat the demonstration of proficiency whenever new staff are trained or
significant changes in instrumentation are made.
9.2.1 Prepare the reference samples from a spiking solution containing each
analyte of interest. The reference sample concentrate (spiking solution) may be prepared
from pure standard materials, or purchased as certified solutions. If prepared by the
laboratory, the reference sample concentrate should be made using stock standards
prepared independently from those used for calibration.

3051A - 8 Revision 1
February 2007
9.2.2 The procedure for preparation of the reference sample concentrate is
dependent upon the method being evaluated. Guidance for reference sample
concentrations for certain methods is listed in Sec. 9.2.4. In other cases, the
determinative methods may contain guidance on preparing the reference sample
concentrate and the reference sample. In the absence of any other guidance, consult
Sec. 9.3.3 and prepare the spiking solution accordingly.
The concentration of target analytes in the reference sample may be adjusted to
more accurately reflect the concentrations that will be analyzed by the laboratory. If the
concentration of an analyte is being evaluated relative to a regulatory limit or action level,
see Sec. 9.3.3 for information on selecting an appropriate spiking level.
9.2.3 To evaluate the performance of the total analytical process, the reference
samples must be handled in exactly the same manner as actual samples. See the note in
Sec. 9.3.1 for important information regarding spiking samples.
9.2.4 Preparation of reference samples for specific determinative methods
The following sections provide guidance on the QC reference sample concentrates
for many determinative methods. The concentration of the target analytes in the QC
reference sample for the methods listed below may need to be adjusted to more
accurately reflect the concentrations of interest in different samples or projects. If the
concentration of an analyte is being evaluated relative to a regulatory limit or action level,
see Sec. 9.3.3 for information on selecting an appropriate spiking level. In addition, the
analyst may vary the concentration of the spiking solution and the volume of solution
spiked into the sample. However, because of concerns about the effects of the spiking
solution solvent on the sample, the total volume spiked into a sample should generally be
held to no more than 1 mL. For any determinative method not listed below, the analyst
should consult Sec. 9.3.3 and is free to choose analytes and spiking concentrations
appropriate for the intended application. See the note in Sec. 9.3.1 for important
information regarding spiking samples.
NOTE
: All of the concentrations listed below refer to the concentration of the spiking
solution itself, not
the concentration of the spiked sample.
9.2.4.1 Method 6010, Inorganic Elements by ICP-AES -- The QC
reference sample concentrate should contain each analyte at 1,000 mg/L in
reagent water with appropriate type(s) and volume(s) of acid(s). See Method
6010.
9.2.4.2 Method 6020, Inorganic Elements by ICP-MS -- The QC
reference sample concentrate should contain each analyte at 1,000 mg/L in
reagent water with appropriate type(s) and volume(s) of acid(s). See Method
6020.
9.2.4.2 Method 7000, Inorganic Elements by Flame AAS -- The QC
reference sample concentrate should contain each analyte at 1,000 mg/L in
reagent water with appropriate type(s) and volume(s) of acid(s). See Method
7000.
9.2.4.3 Method 7010, Inorganic Elements by Graphite Furnace AAS --
The QC reference sample concentrate should contain each analyte at 1,000 mg/L
3051A - 9 Revision 1
February 2007
in reagent water with appropriat type(s) and volume(s) of acid(s). See Method
7010.
9.2.4.4 Method 7061, Arsenic by AA, Gaseous Hydride -- The QC
reference sample concentrate should contain arsenic at 1,000 mg/L in reagent
water with appropriate volume of concentrated nitric acid. See Method 7061.
9.2.4.5 Method 7062, Antimony and Arsenic by AA, Borohydride
Reduction -- The QC reference sample concentrate should contain each analyte at
1,000 mg/L in reagent water with appropriate volume of concentrated nitric acid.
See Method 7062.
9.2.4.5 Method 7063, Arsenic by ASV -- The QC reference sample
concentrate should contain mercury at 1,000 mg/L in reagent water with
appropriate volume of concentrated nitric acid. Stock solutions are commercially
available as spectrophotometric standards. See Method 7063.
9.2.4.6 Method 7470, Mercury in Liquid Waste by Manual Cold-Vapor
Technique -- The QC reference sample concentrate should contain mercury at
1,000 mg/L in reagent water with appropriate volume of concentrated nitric acid.
Stock solutions are also commercially available as spectrophotometric standards.
See Method 7470.
9.2.4.7 Method 7471, Mercury in Solid or Semisolid Waste by Manual
Cold-Vapor Technique -- The QC reference sample concentrate should contain
mercury at 1,000 mg/L in reagent water with appropriate volume of concentrated
nitric acid. Stock solutions are also commercially available as spectrophotometric
standards. See Method 7471.
9.2.4.8 Method 7472, Mercury by ASV -- The QC reference sample
concentrate should contain mercury at 1,000 mg/L in reagent water with
appropriate volume of concentrated nitric acid. Stock solutions are also
commercially available as spectrophotometric standards. See Method 7472.
9.2.4.9 Method 7473, Mercury by Thermal, Decomposition,
Amalgamation, and AA -- The QC reference sample concentrate should contain
mercury at 1,000 mg/L in reagent water with appropriate volume of concentrated
nitric acid. Stock solutions are also commercially available as spectrophotometric
standards. See Method 7473.
9.2.4.10 Method 7474, Mercury by Atomic Fluorescence -- The QC
reference sample concentrate should contain mercury at 1,000 mg/L in reagent
water with appropriate volume of concentrated nitric acid. Stock solutions are also
commercially available as spectrophotometric standards. See Method 7474.
9.2.4.11 Method 7741, Selenium by AA, Gaseous Reduction -- The QC
reference sample concentrate should contain selenium at 1,000 mg/L in reagent
water. See Method 7741.
9.2.4.12 Method 7742, Selenium by AA, Borohydride Reduction -- The
QC reference sample concentrate should contain selenium at 1,000 mg/L in
reagent water. See Method 7742.

3051A - 10 Revision 1
February 2007
9.2.5 Analyze at least four replicate aliquots of the well-mixed reference
samples by the same procedures used to analyze actual samples. This will include a
combination of the sample preparation method and the determinative method (a 6000 or
7000 series method). Follow the guidance on data calculation and interpretation
presented in the determinative method.
9.3 Sample quality control for preparation and analysis
9.3.1 Documenting the effect of the matrix should include the analysis of at
least one matrix spike and one duplicate unspiked sample or one matrix spike/matrix spike
duplicate pair per analytical batch. The decision on whether to prepare and analyze
duplicate samples or a matrix spike/matrix spike duplicate must be based on a knowledge
of the samples in the sample batch. If samples are expected to contain target analytes,
laboratories may use one matrix spike and a duplicate analysis of an unspiked field
sample. If samples are not expected to contain target analytes, then laboratories should
use a matrix spike and matrix spike duplicate pair. The consideration as to which sample
for a given batch is selected for QC analyses should be decided during the project
planning process and documented in an approved sampling and analysis plan. The actual
sample selected for QC analyses should be representative of the entire matrix composition
for a given extraction batch, since data quality assumptions will likely be applied to all
batch samples based on compliance to the stated data quality objectives and meeting the
recommended precision and accuracy criteria. Therefore, it is inappropriate to combine
dissimilar matrices in a single sample preparatory batch and expect to use a single set of
QC samples. Sec. 9.3.3 provides guidance on establishing the concentration of the matrix
spike compounds in the sample chosen for spiking.
The choice of analytes to be spiked should reflect the analytes of interest for the
specific project. Thus, if only a subset of the list of target analytes provided in a
determinative method are of interest, then these would be the analytes of interest for the
project. In the absence of project-specific analytes of interest, it is suggested that the
laboratory periodically change the analytes that are spiked with the goal of obtaining
matrix spike data for most, if not all, of the analytes in a given determinative method. If
these compounds are not target analytes for a specific project, or if other compounds are
known to be of greater concern at a given site, then other matrix spike compounds should
be employed.
CAUTION:
The utility of the data for the matrix spike compounds depends on the degree
to which the spiked compounds mimic the compounds already present in a
field sample. Therefore, it is CRITICAL that any compounds added to a
sample are added to the sample aliquot PRIOR TO any additional processing
steps. It is also CRITICAL that the spiked compounds be in the same
chemical form as the target compounds.
9.3.2 A laboratory control sample (LCS) should be included with each analytical
batch. The LCS consists of an aliquot of a clean (control) matrix similar to the sample
matrix and of the same weight or volume: e.g., reagent water for the water matrix or sand
or soil for the solid matrix. The LCS is spiked with the same analytes at the same
concentrations as the matrix spike, when appropriate. When the results of the matrix
spike analysis indicate a potential problem due to the sample matrix itself, the LCS results
are used to verify that the laboratory can perform the analysis in a clean matrix.
9.3.3 The concentration of the matrix spike sample and/or the LCS should be
determined as described in the following sections.
3051A - 11 Revision 1
February 2007
9.3.3.1 If, as in compliance monitoring, the concentration of a specific
analyte in the sample is being checked against a regulatory limit or action level, the
spike should be at or below the regulatory limit or action level, or 1 - 5 times the
background concentration (if historical data are available), whichever concentration
is higher.
9.3.3.2 If historical data are not available, it is suggested that an
uncontaminated sample of the same matrix from the site be submitted for matrix
spiking purposes to ensure that high concentrations of target analytes and/or
interferences will not prevent calculation of recoveries.
9.3.3.3 If the concentration of a specific analyte in a sample is not
being checked against a limit specific to that analyte, then the concentration of the
matrix spike should be at the same concentration as the reference sample (Sec.
9.2.4), near the middle of calibration range, or approximately 10 times the
quantitation limit in the matrix of interest. It is again suggested that a background
sample of the same matrix from the site be submitted as a sample for matrix
spiking purposes.
9.3.4 Analyze these QC samples (the LCS and the matrix spikes or the optional
matrix duplicates) following the procedures in the determinative method. Calculate and
evaluate the QC data as outlined in the determinative method.
9.3.5 Blanks -- The preparation and analysis of method blanks and other blanks
are necessary to track potential contamination of samples during the extraction and
analysis processes. Refer to Chapter One for specific quality control procedures.
9.4 The laboratory must also have procedures for documenting the effect of the matrix
on method performance. Refer to Chapter One and each determinative method for specific
guidance on developing method performance data.
9.5 Periodically, the accuracy of the temperature measurement system used to control
the microwave equipment should be validated per Sec. 6.1.2.
9.6 (This step is not necessary if using temperature feedback control.) Each day that
samples are extracted, the microwave-power calibration should be verified by heating 1 kg of
ASTM Type II water (at 22 ± 3 EC) in a covered, microwave-transparent vessel for 2 min at the
setting for 490 W and measuring the water temperature after heating per Sec. 10.5. If the
power calculated (according to Sec. 12.0) differs from 490 W by more than ± 10 W, the
microwave settings should be recalibrated according to Sec. 10.0.
9.7 The choice of an acid or acid mixture for digestion will depend on the analytes of
interest and no single acid or acid mixture is universally applicable to all analyte groups.
Whatever acid or acid mixture is employed, including those specifically listed in this method, the
analyst must demonstrate adequate performance for the analytes of interest, at the levels of
interest. At a minimum, such a demonstration will encompass the initial demonstration of
proficiency described in Method 3500, using a clean reference matrix. Method 8000 describes
procedures that may be used to develop performance criteria for such demonstrations as well
as for matrix spike and laboratory control sample results.

3051A - 12 Revision 1
February 2007
10.0 CALIBRATION AND STANDARDIZATION
The following sections provide information regarding the calibration of microwave
equipment.
NOTE
: If the microwave unit uses temperature feedback control to control the performance
specifications of the method, then performing the calibration procedure is not
necessary.
10.1 Calibration is the normalization and reproduction of a microwave field strength to
permit reagent and energy coupling in a predictable and reproducible manner. It balances
reagent heating and heat loss from the vessels and is equipment dependent due to the heat
retention and loss characteristics of the specific vessel. Available power is evaluated to permit
the microwave field output in watts to be transferred from one microwave system to another.
Use of calibration to control this reaction requires balancing output power, coupled energy,
and heat loss to reproduce the temperature heating profile given in Sec. 11.3.5. The conditions
for each acid mixture and each batch containing the same specified number of vessels must be
determined individually. Only identical acid mixtures and vessel models and specified numbers
of vessels may be used in a given batch.
10.2 For cavity type microwave equipment, calibration is accomplished by measuring
the temperature rise in 1 kg of water exposed to microwave radiation for a fixed period of time.
The analyst can relate power in watts to the partial power setting of the system. The calibration
format needed for laboratory microwave systems depends on the type of electronic system used
by the manufacturer to provide partial microwave power. Few systems have an accurate and
precise linear relationship between percent power settings and absorbed power. Where linear
circuits have been utilized, the calibration curve can be determined by a three-point calibration
method (see Sec. 10.4). Otherwise, the analyst must use the multiple point calibration method
(see Sec. 10.3). Assistance in calibration and software guidance of calibration are available in
Ref. 3 and the document listed in Sec. 13.3.5.
10.3 The multiple point calibration involves the measurement of absorbed power over a
large range of power settings. Typically, for a 600 W unit, the following power settings are
measured: 100, 99, 98, 97, 95, 90, 80, 70, 60, 50, and 40% using the procedure described in
Sec. 10.5. These data are clustered about the customary working power ranges. Non-linearity
has been encountered at the upper end of the calibration. Non-linearity is primarily encountered
when using older instrumentation, however, multi-point calibration is recommended for use with
all instrumentation when accurate and precise temperature feedback control is not available. If
the system's electronics are known to have nonlinear deviations in any region of proportional
power control, it will be necessary to make a set of measurements that bracket the power to be
used. The final calibration point should be at the partial power setting that will be used in the
test. This setting should be checked periodically to evaluate the integrity of the calibration. If a
significant change is detected (± 10 W), then the entire calibration should be re-evaluated.
10.4 The three-point calibration involves the measurement of absorbed power at three
different power settings. Measure the power at 100% and 50% using the procedure described
in Sec. 10.5. From this 2-point line, determine the partial power setting that corresponds to the
power, in watts, specified in the procedure to reproduce the heating profile specified in Sec.
11.3.5. Measure the absorbed power at that partial power setting. If the measured absorbed
power does not correspond to the specified power within ± 10 W, use the multiple point
calibration in Sec. 10.3. This point should also be used to periodically verify the integrity of the
calibration.

3051A - 13 Revision 1
February 2007
10.5 Equilibrate a large volume of water to room temperature (22 ± 3 EC). One kg of
reagent water is weighed (1,000.0 ± 0.1 g) into a fluorocarbon beaker or a beaker made of
some other material that does not significantly absorb microwave energy (glass absorbs
microwave energy and is not recommended). The initial temperature of the water should be 22
± 3 EC measured to ± 0.05 EC. The covered beaker is circulated continuously (in the normal
sample path) through the microwave field for 2 min at the desired partial power setting with the
system's exhaust fan on maximum (as it will be during normal operation). The beaker is
removed and the water vigorously stirred. Use a magnetic stirring bar inserted immediately
after microwave irradiation (irradiating with the stir bar in the vessel could cause electrical
arcing) and record the maximum temperature within the first 30 seconds to ± 0.05 EC. Use a
new sample for each additional measurement. If the water is reused (after making adjustments
for any loss of weight due to heating), both the water and the beaker must have returned to 22 ±
3 EC. Three measurements at each power setting should be made.
The absorbed power is determined by the following relationship:
P'
(K)(C
p
)(m)(∆T)
t
Where:
P = the apparent power absorbed by the sample in watts (W) (joule/sec)
K = the conversion factor for thermochemical calories sec
-1
to watts (K= 4.184)
C
p
= the heat capacity, thermal capacity, or specific heat [cal/(g EC)] of water
m = the mass of the water sample in grams (g)
∆T = the final temperature minus the initial temperature (EC)
t = the time in seconds (s)
Using the experimental conditions of 2 minutes (120 sec) and 1 kg (1000 g) of distilled
water [heat capacity at 25 EC is 0.9997 cal/(g EC)], the calibration equation simplifies to:
P'
∆T 34.86
NOTE:
Stable line voltage is necessary for accurate and reproducible calibration and
operation. The line voltage should be within manufacturer's specification, and during
measurement and operation should not vary by more than ± 2 V (Ref. 3). Electronic
components in most microwave units are matched to the system's function and output.
When any part of the high voltage circuit, power source, or control components in the
system are serviced or replaced, it will be necessary to recheck the system’s

3051A - 14 Revision 1
February 2007
calibration. If the power output has changed significantly (± 10 W), then the entire
calibration should be re-evaluated.
11.0 PROCEDURE
11.1 Temperature control of closed vessel microwave instruments provides the main
feedback control performance mechanism for this method. Method control requires a
temperature sensor in one or more vessels during the entire decomposition. The microwave
decomposition system should sense the temperature to within ± 2.5 EC and permit adjustment
of the microwave output power within 2 sec.
11.2 All digestion vessels and volumetric ware must be carefully acid washed and
rinsed with reagent water. When switching between high concentration samples and low
concentration samples, all digestion vessels (fluoropolymer or quartz liners) should be cleaned
by leaching with hot (1:1) hydrochloric acid (greater than 80 EC, but less than boiling) for a
minimum of two hours followed by hot (1:1) nitric acid (greater than 80 EC, but less than boiling)
for a minimum of two hours. The vessels should then be rinsed with reagent water and dried in
a clean environment. This cleaning procedure should also be used whenever the prior use of
the digestion vessels is unknown or cross contamination from prior sample digestions in vessels
is suspected. Polymeric or glass volumetric ware and storage containers should be cleaned by
leaching with more dilute acids (approximately 10% V/V) appropriate for the specific material
used and then rinsed with reagent water and dried in a clean environment.
11.3 Sample digestion
11.3.1 Weigh a well-mixed sample to the nearest 0.001 g into an appropriate
vessel equipped with a controlled pressure relief mechanism. For soils, sediments, and
sludges, use no more than 0.500 g. For oil or oil contaminated soils, initially use no more
than 0.250 g. When large samples of oil are necessary, the use of Method 3052, which
has sample scale-up options, is recommended. If the sample can not be well mixed and
homogenized on an as received basis, then perform air or oven drying at 60 °C or less,
crushing, sieving, grinding, and mixing as necessary to homogenize the sample until the
subsampling variance is less than the data quality objectives of the analysis. While proper
sample preparation generally produces great reduction in analytical variability, be aware
that in certain unusual circumstances there could be loss of volatile metals (e.g., Hg,
organometallics) or irreversible chemical changes ( e.g., precipitation of insoluble species,
change in valence state).
11.3.2 Add 10 ± 0.1 mL concentrated nitric acid or, alternatively, 9 ± 0.1 mL
concentrated nitric acid and 3 ± 0.1 mL concentrated hydrochloric acid to the vessel in a
fume hood (or fume exhausted enclosure). The addition of concentrated hydrochloric acid
to the nitric acid is appropriate for the stabilization of certain analytes, such as Ag, Ba, and
Sb and high concentrations of Fe and Al in solution. Improvements and optimal
recoveries of antimony, iron, and silver from a variety of matrices upon addition of HCl are
demonstrated in Sec. 17.0, in Figures 3 through 7 (these data are provided for guidance
purposes only). The addition of hydrochloric acid may, however, limit the quantitation
techniques or increase the difficulties of analysis for some quantitation systems.
WARNING
: The addition of hydrochloric acid must be in the form of concentrated
hydrochloric acid and not from a premixed combination of acids as a buildup
of chlorine gas, as well as other gases, will result from a premixed acid

3051A - 15 Revision 1
February 2007
solution. These gases may be violently released upon heating. This is
avoided by adding the acid in the described manner.
WARNING
: Toxic nitrogen oxide(s) and chlorine fumes are usually produced during
digestion. Therefore, all steps involving open or the opening of microwave
vessels must be performed in a properly operating fume ventilation system.
WARNING
: The analyst should wear protective gloves and face protection.
CAUTION
: The use of microwave equipment with temperature feedback control is
needed to control any unfamiliar reactions that may occur during the leaching
of samples of unknown composition. The leaching of these samples may
require additional vessel requirements such as increased pressure
capabilities.
11.3.3 The analyst should be aware of the potential for a vigorous reaction,
especially with samples containing volatile or easily oxidized organic species. When
digesting a matrix of this type, do not leach this type of sample as described in this
method, due to the high potential for unsafe and uncontrollable reactions. Instead, these
samples may be predigested in a hood, with the vessel loosely capped to allow gases to
escape, eliminating the hazard presented by rapid addition of thermal energy (MW power)
to a reactive mixture. After predigestion, the samples may be digested according to the
procedures described in this method.
11.3.4 Seal the vessel according to the manufacturer's directions. Properly
place the vessel in the microwave system according to the manufacturer's recommended
specifications and, when applicable, connect appropriate temperature and pressure
sensors to vessels according to manufacturer’s specifications.
11.3.5 This method is compatible with a performance-based approach, designed
to achieve or approach consistent leaching of the sample through achieving specific
reaction conditions. The temperature of each sample should rise to 175 ± 5 EC in
approximately 5.5 ± 0.25 min and remain at 175 ± 5 EC for 4.5 min, or for the remainder of
the 10-min digestion period (see Refs. 2, 3, and 4 and the document listed in Sec. 13.3.4).
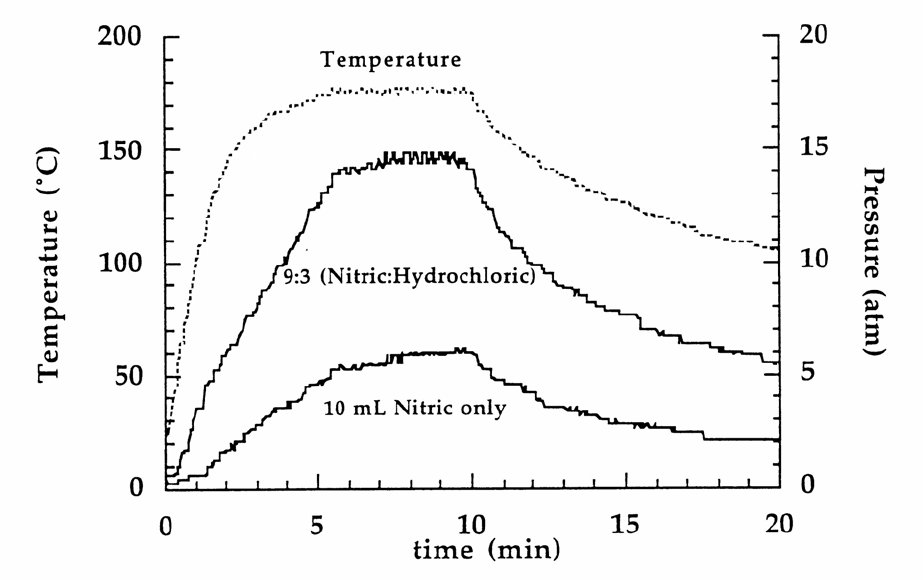
Figure 2 gives the time versus temperature and pressure profile for a standard sediment
sample (these data are presented for guidance purposes only). When using temperature
feedback control, the number of samples that may be simultaneously digested may vary,
from one sample up to the maximum number of vessels that can be heated by the
magnetron of the microwave unit according to the heating profile specified previously in
this section. The number will depend on the power of the unit, the number of vessels, and
the heat loss characteristics of the vessels (Ref. 3).
11.3.5.1 The pressure should peak between 5 and 10 min for most
samples (see Refs. 1 and 2 and the document listed in Sec. 13.3.4). If the
pressure exceeds the pressure limits of the vessel, the pressure should be safely
and controllably reduced by the pressure relief mechanism of the vessel.
11.3.5.2 Calibration control is applicable in reproducing this method
provided the power in watts versus time parameters are determined to reproduce
the specifications listed in Sec. 11.3.5. The calibration settings will be specific to
the quantity of reagents, the number of vessels, and the heat loss characteristics of
the vessels (see Ref. 3 and the document listed in Sec. 13.3.3). If calibration
control is being used, any vessels containing acids for analytical blank purposes
3051A - 16 Revision 1
February 2007
are counted as sample vessels. When fewer than the recommended number of
samples are to be digested, the remaining vessels should be filled with the same
acid mixture to achieve the full complement of vessels. This provides an energy
balance, since the microwave power absorbed is proportional to the total absorbing
mass in the cavity. Irradiate each group of vessels using the predetermined
calibration settings. (Different vessel types should not be mixed.)
11.3.6 At the end of the microwave program, allow the vessels to cool for a
minimum of 5 min before removing them from the microwave system. Cooling of the
vessels may be accelerated by internal or external cooling devices. When the vessels
have cooled to near room temperature, determine if the microwave vessels have
maintained their seal throughout the digestion. Due to the wide variability of vessel
designs, a single procedure is not appropriate. For vessels that are sealed as discrete
separate entities, the vessel weight may be taken before and after digestion to evaluate
seal integrity. If the weight loss of sample exceeds 1% of the weight of the sample and
reagents, then the sample is considered compromised. For vessels with burst disks, a
careful visual inspection of the disk, in addition to weighing, may identify compromised
vessels. For vessels with resealing pressure relief mechanisms, an auditory or a physical
sign that can indicate whether a vessel has vented is appropriate.
11.3.7 Complete the preparation of the sample by venting microwave containers
in a fume hood before uncapping, so as to avoid a rush of acid vapor that may still be in
the headspace. When sufficiently cool to handle, carefully uncap the vessels, using the
procedure recommended by the vessel manufacturer. Quantitatively transfer the sample
to an acid-cleaned bottle. If the digested sample contains particulates which may clog
nebulizers or interfere with injection of the sample into the instrument, the sample may be
centrifuged (Sec. 11.3.7.1), allowed to settle (Sec. 11.3.7.2), or filtered (Sec. 11.3.7.3).
11.3.7.1 Centrifugation -- Centrifugation at 2,000 - 3,000 rpm for 10 min
is usually sufficient to clear the supernatant.
11.3.7.2 Settling -- If undissolved material, such as SiO
2
, TiO
2
, or other
refractory oxides, remains, allow the sample to stand until the supernatant is clear.
Allowing a sample to stand overnight will usually accomplish this. If it does not,
centrifuge or filter the sample.
11.3.7.3 Filtering -- If necessary, the filtering apparatus must be
thoroughly cleaned and pre-rinsed with dilute (approximately 10% V/V) nitric acid.
Filter the sample through qualitative filter paper into a second acid-cleaned
container.
11.3.8 The removal or reduction of the quantity of the nitric and hydrochloric
acids prior to analysis may be desirable. The chemistry and volatility of the analytes of
interest should be considered and evaluated when using this alternative (Ref. 3).
Evaporation to near dryness in a controlled environment with controlled purge gas and
neutralizing and collection of exhaust interactions is an alternative where appropriate.
This manipulation may be performed in the microwave system, if the system is capable of
this function, or external to the microwave system in more common apparatus(s). This
option must be tested and validated to determine analyte retention and loss and should be
accompanied by equipment validation possibly using the standard addition method and
standard reference materials. This alternative may be used to alter either the acid
concentration and/or acid composition prior to analysis. (For further information, see Ref.
3 and Method 3052).

3051A - 17 Revision 1
February 2007
NOTE
: The final solution typically requires nitric acid to maintain appropriate sample
solution acidity and stability of the elements. Commonly, a 2% (v/v) nitric acid
concentration is desirable. Waste minimization techniques should be used to
capture reagent fumes. This procedure should be tested and validated in the
apparatus and on standards before using on unknown samples.
11.3.9 Transfer or decant the sample into volumetric ware and dilute the digest to a
known volume. The digest is now ready for analysis for elements of interest using appropriate
elemental analysis techniques.
12.0 DATA ANALYSIS AND CALCULATIONS
12.1 Calculations -- The concentrations determined are to be reported on the basis of
the actual weight of the original sample. All dilutions must be taken into account when
computing the final results.
12.2 Prior to using this method, verify that the temperature sensing equipment is
properly reading temperature. A procedure for verification is given in Sec. 6.1.2. This will
establish the accuracy and precision of the temperature sensing equipment, which should be
carried throughout the statistical treatment of the quality assurance data.
12.3 In calibrating the microwave unit (Sec. 10.0), the power absorbed (for each power
setting) by 1 kg of reagent water exposed to 120 seconds of microwave energy is determined by
the expression
Power (in watts) = (T
1
- T
2
) (34.86)
Where:
T
1
= Initial temperature of water (between 21 and 25 EC to nearest 0.1 EC)
T
2
= Final temperature of water (to nearest 0.1 EC)
12.4 Plot the power settings against the absorbed power (calculated in Sec. 12.3) to
obtain a calibration relationship. Alternatively, use a microwave calibration program to analyze
the calibration data (see Ref. 3 and the document listed in Sec. 13.3.5). Interpolate the data to
obtain the instrument settings needed to provide the wattage levels specified in Sec. 12.3.
12.5 Calculate the sample dry-weight fraction as follows:
Dry-Wt fraction =
W
2
& W
3
W
1
& W
3
Where:
W
1
= Wt for sample + vessel, before drying, g

3051A - 18 Revision 1
February 2007
W
2
= Wt for sample + vessel, after drying, g
W
3
= Wt for empty, dry vessel, g
12.6 Convert the extract concentration obtained from the instrument in mg/L to mg/kg
dry-weight of sample by:
Sample concentration =
C V D
W S
Where:
C = Concentration in extract (mg/L)
D = Dilution factor
S = Solid dry-weight fraction for sample, g/g
V = Volume of extract, mL x 0.001
W = Weight of undried sample extracted, g x 0.001
13.0 METHOD PERFORMANCE
13.1 Performance data and related information are provided in SW-846 methods only as
examples and guidance. The data do not represent required performance criteria for users of
the methods. Instead, performance criteria should be developed on a project-specific basis,
and the laboratory should establish in-house QC performance criteria for the application of this
method. These performance data are not intended to be and must not be used as absolute QC
acceptance criteria for purposes of laboratory accreditation.
13.2 The fundamental chemical basis of this method with and without HCl has been
compared with Method 3050 in several sources (see 13.3.4 and 13.3.6). Several papers have
evaluated the leachability of NIST SRMs with this method (Ref. 1 and Sec. 13.3.5). Evaluations
and optimizations of this method have been documented (Refs. 5 and 6), as well as additional
leaches performed on more matrices, which may be addressed in future papers. This method
has been determined to be appropriate for enhancing recoveries of certain analytes. This data
is contained in Sec. 17 of this method. Matrices tested include SRM 2710 (Montana Soil -
Highly Elevated Concentrations), SRM 2704 (Buffalo River Sediment), and SRM 1084a (Wear
Metals in Oil). Analytes demonstrating better recoveries upon addition of HCl include antimony,
iron, and silver. These data are provided for guidance purposes only.
13.3 The following documents may provide additional guidance and insight on this
method and technique:
13.3.1 H. M. Kingston and L. B. Jassie, "Safety Guidelines for Microwave
Systems in the Analytical Laboratory," in Introduction to Microwave Acid Decomposition:
Theory and Practice, Kingston, H.M. and Jassie, L.B., eds., ACS Professional Reference
Book Series, American Chemical Society, Washington, DC, 1988.

3051A - 19 Revision 1
February 2007
13.3.2 1985 Annual Book of ASTM Standards
, Vol. 11.01; "Standard
Specification for Reagent Water," ASTM, Philadelphia, PA, 1985, D1193-77.
13.3.3 Introduction to Microwave Sample Preparation: Theory and Practice
,
Kingston, H. M. and Jassie, L. B., Eds., ACS Professional Reference Book Series,
American Chemical Society, Washington, DC, 1988.
13.3.4 H. M. Kingston and P. J. Walter, "Comparison of Microwave Versus
Conventional Dissolution for Environmental Applications," Spectroscopy, Vol. 7 No. 9, 20-
27, 1992.
13.3.5 P. J. Walter, Special Publication IR4718: Microwave Calibration
Program, 2.0 ed., National Institutes of Standards and Technology, Gaithersburg, MD,
1991.
13.3.6 H. M. Kingston, P. J. Walter, S. J. Chalk, E. M. Lorentzen, D. D. Link,
"Environmental Microwave Sample Preparation: Fundamentals, Methods, and
Applications," in Microwave Enhanced Chemistry: Fundamentals, Sample Preparation,
and Applications, ACS Professional Reference Book Series, American Chemical Society,
Washington, DC 1997.
14.0 POLLUTION PREVENTION
14.1 Pollution prevention encompasses any technique that reduces or eliminates the
quantity or toxicity of waste at the point of generation. Numerous opportunities for pollution
prevention exist in laboratory operations. The EPA has established a preferred hierarchy of
environmental management techniques that places pollution prevention as the management
option of first choice. Whenever feasible, laboratory personnel should use pollution prevention
techniques to address their waste generation. When wastes cannot be feasibly reduced at the
source, the Agency recommends recycling as the next best option.
14.2 For information about pollution prevention that may be applicable to laboratories
and research institutions consult Less is Better: Laboratory Chemical Management for Waste
Reduction available from the American Chemical Society's Department of Government
Relations and Science Policy,1155 16th Street, NW, Washington, DC 20036,
http://www.acs.org
.
15.0 WASTE MANAGEMENT
The Environmental Protection Agency requires that laboratory waste management
practices be consistent with all applicable rules and regulations. The Agency urges laboratories
to protect the air, water, and land by minimizing and controlling all releases from hoods and
bench operations, complying with the letter and spirit of any sewer discharge permits and
regulations, and by complying with all solid and hazardous waste regulations, particularly the
hazardous waste identification rules and land disposal restrictions. For further information on
waste management, consult The Waste Management Manual for Laboratory Personnel
,
available from the American Chemical Society's Department of Government Relations and
Science Policy, 1155 16th Street, NW, Washington, DC 20036, (202) 872-4477.

3051A - 20 Revision 1
February 2007
16.0 REFERENCES
1. H. M. Kingston, EPA IAG #DWI-393254-01-0 January 1 - March 31, 1988, quarterly report.
2. D. A. Binstock, W. M. Yeager, P. M. Grohse and A. Gaskill, "Validation of a Method for
Determining Elements in Solid Waste by Microwave Digestion," Research Triangle
Institute Technical Report Draft, RTI Project Number 321U-3579-24, November, 1989,
prepared for the Office of Solid Waste, U.S. Environmental Protection Agency,
Washington, DC 20460.
3. H. M. Kingston, S. Haswell, Microwave Enhanced Chemistry: Fundamentals, Sample
Preparation, and Applications, ACS Professional Reference Book Series, American
Chemical Society, Washington, DC 1997.
4. D. A. Binstock, P. M. Grohse, A. Gaskill, C. Sellers, H. M. Kingston, L. B. Jassie,
"Development and Validation of a Method for Determining Elements in Solid Waste Using
Microwave Digestion," J. Assoc. Off. Anal. Chem., Vol. 74, 360 - 366, 1991.
5. H. M. Kingston, P. J. Walter, E. M., L. Lorentzen, G. P. Lusnak, "The Performance of
Leaching Studies on Soil SRM’s 2710 and 2711," Final Report to the National Institute of
Standards and Technology, Duquesne University, Pittsburgh, PA, April 5, 1994.
6. D. D. Link, H. M. Kingston, P. J. Walter, "Development and Validation of the New EPA
Microwave-assisted Leach Method 3051A," Environmental Science and Technology, Vol.
32, p. 3628-3632, 1998.
7. D. D. Link, H. M. Kingston, P. J. Walter, "Development and Validation of the EPA
Microwave-assisted Methods 3015A and 3051A: Validation Studies for Updated
Microwave Leach Methods," Proceedings for the Waste Testing and Quality Assurance
Symposium, July 1997.
8. H. M. Kingston, P. J. Walter, "Comparison of Microwave verses Conventional Dissolution
for Environmental Applications," Spectroscopy, Vol. 7 No. 9, 20-27, 1992.
17.0 TABLES, DIAGRAMS, FLOWCHARTS, AND VALIDATION DATA
The following pages contain the tables and figures referenced by this method.

3051A - 21 Revision 1
February 2007
TABLE 1
COMPARISON OF ANALYTE RECOVERIES FROM SRM 2704 (BUFFALO RIVER
SEDIMENT)
USING BOTH DIGEST OPTIONS
Element 10 mL HNO
3
digest
9 mL HNO
3
+
3 mL HCl digest
Total Analyte
Concentration
Cd 3.40 ± 0.34 3.62 ± 0.17 3.45 ± 0.22
Cr 84.7 ± 5.6 77.1 ± 12.6 135 ± 5
Ni 45.5 ± 5.9 42.2 ± 3.2 44.1 ± 3.0
Pb 163 ± 9 161 ± 17 161 ± 17
Elemental analysis was performed using either FAAS or ICP-MS.
Results reported in µg/g analyte (mean ± 95% confidence limit).
Total concentrations are taken from NIST SRM Certificate of Analysis.
These data are provided for guidance purposes only.
Data taken from Refs. 6 and 7.
TABLE 2
COMPARISON OF ANALYTE RECOVERIES FROM SRM 4355 (PERUVIAN SOIL)
USING BOTH DIGEST OPTIONS
Element 10 mL HNO
3
digest
9 mL HNO
3
+
3 mL HCl digest
Total Analyte
Concentration
Cd 0.86 ± 0.16 0.85 ± 0.17 (1.50)
Cr 14.6 ± 0.47 19.0 ± 0.69 28.9 ± 2.8
Ni 9.9 ± 0.33 11.2 ± 0.44 (13)
Pb 124 ± 5.3 130 ± 3.6 129 ± 26
Elemental analysis was performed using either FAAS or ICP-MS.
Results reported in µg/g analyte (mean ± 95% confidence limit).
Total concentrations are taken from NIST SRM Certificate of Analysis.
Values in parenthesis are reference concentrations.
These data are provided for guidance purposes only.
Data taken from Refs. 6 and 7.

3051A - 22 Revision 1
February 2007
TABLE 3
COMPARISON OF ANALYTE RECOVERIES FROM SRM 1084a (WEAR METALS IN OIL)
USING BOTH DIGEST OPTIONS
Element 10 mL HNO
3
digest
9 mL HNO
3
+
3 mL HCl digest
Total Analyte
Concentration
Cu 91.6 ± 4.0 93.0 ± 2.6 100.0 ± 1.9
Cr 91.2 ± 3.3 94.3 ± 3.1 98.3 ± 0.8
Mg 93.2 ± 3.6 93.5 ± 2.8 99.5 ± 1.7
Ni 91.6 ± 3.9 92.9 ± 3.4 99.7 ± 1.6
Pb 104 ± 4.1 99.5 ± 5.1 101.1 ± 1.3
Elemental analysis was performed using either FAAS or ICP-MS.
Results reported in µg/g analyte (mean ± 95% confidence limit).
Total concentrations are taken from NIST SRM Certificate of Analysis.
These data are provided for guidance purposes only.
Data taken from Refs. 6 and 7.

3051A - 23 Revision 1
February 2007
FIGURE 1
PRESSURE PROFILES FOR THE HEATING OF DIFFERENT RATIOS
OF NITRIC ACID TO HYDROCHLORIC ACID
Figure taken from Refs. 6 and 7.
3051A - 24 Revision 1
February 2007
FIGURE 2
TEMPERATURE AND PRESSURE PROFILE
FOR NIST SRM 2704 (BUFFALO RIVER SEDIMENT)
USING DIFFERENT RATIOS OF NITRIC ACID TO HYDROCHLORIC ACID
Figure taken from Refs. 6 and 7.

3051A - 25 Revision 1
February 2007
FIGURE 3
PERCENT RECOVERY OF ANTIMONY FROM NIST SRM 2710 (MONTANA SOIL) VERSUS
VARIOUS COMBINATIONS OF NITRIC AND HYDROCHLORIC ACIDS (N=6)
Figure taken from Refs. 6 and 7.

3051A - 26 Revision 1
February 2007
FIGURE 4
PERCENT RECOVERY OF ANTIMONY FROM NIST SRM 2704 (BUFFALO RIVER SEDIMENT)
VERSUS VARIOUS COMBINATIONS OF NITRIC AND HYDROCHLORIC ACIDS(N=6)
Figure taken from Refs. 6 and 7.

3051A - 27 Revision 1
February 2007
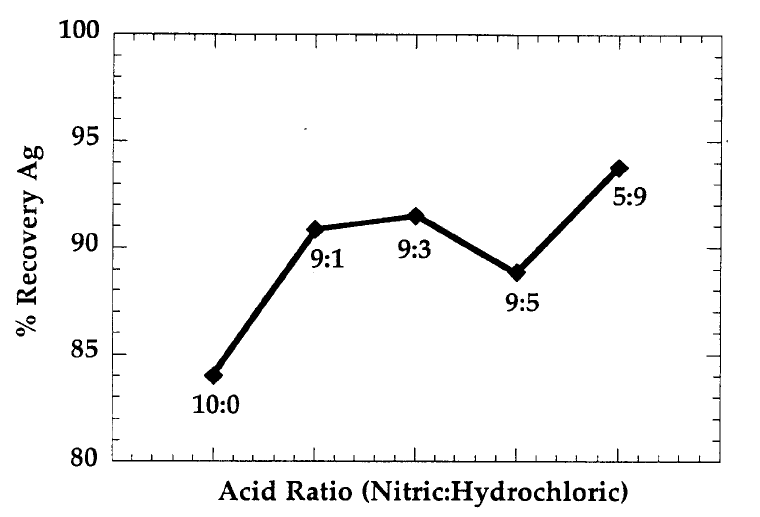
FIGURE 5
PERCENT RECOVERY OF IRON FROM NIST SRM 2704 (BUFFALO RIVER SEDIMENT)
VERSUS VARIOUS COMBINATIONS OF NITRIC AND HYDROCHLORIC ACIDS (N=6)
Figure taken from Refs. 6 and 7.
3051A - 28 Revision 1
February 2007
FIGURE 6
PERCENT RECOVERY OF SILVER FROM NIST SRM 2710 (MONTANA SOIL) VERSUS
VARIOUS COMBINATIONS OF NITRIC AND HYDROCHLORIC ACIDS (N=6)
Figure taken from Refs. 6 and 7.

3051A - 29 Revision 1
February 2007
FIGURE 7
PERCENT RECOVERY OF ANTIMONY AND IRON, RESPECTIVELY, FROM SRM 4355
(PERUVIAN SOIL) USING BOTH DIGEST OPTIONS
(10 ML HNO
3
AND 9 ML HNO
3
+ 3 ML HCL ) (N=6)
Figure taken from Refs. 6 and 7.

3051A - 30 Revision 1
February 2007
METHOD 3051A
MICROWAVE ASSISTED ACID DIGESTION OF SEDIMENTS, SLUDGES, SOILS, AND OILS
